Ein einheitlicher ISO-Standard zur Identifikation von Arzneimitteln «Identification of Medicinal Products» soll weltweit Anwendung finden – im EMA-Raum bereits ab diesem Jahr. Mit mehreren Jahren Abstand will auch die Swissmedic folgen. Was bedeutet das für Zulassungsinhaberinnen?
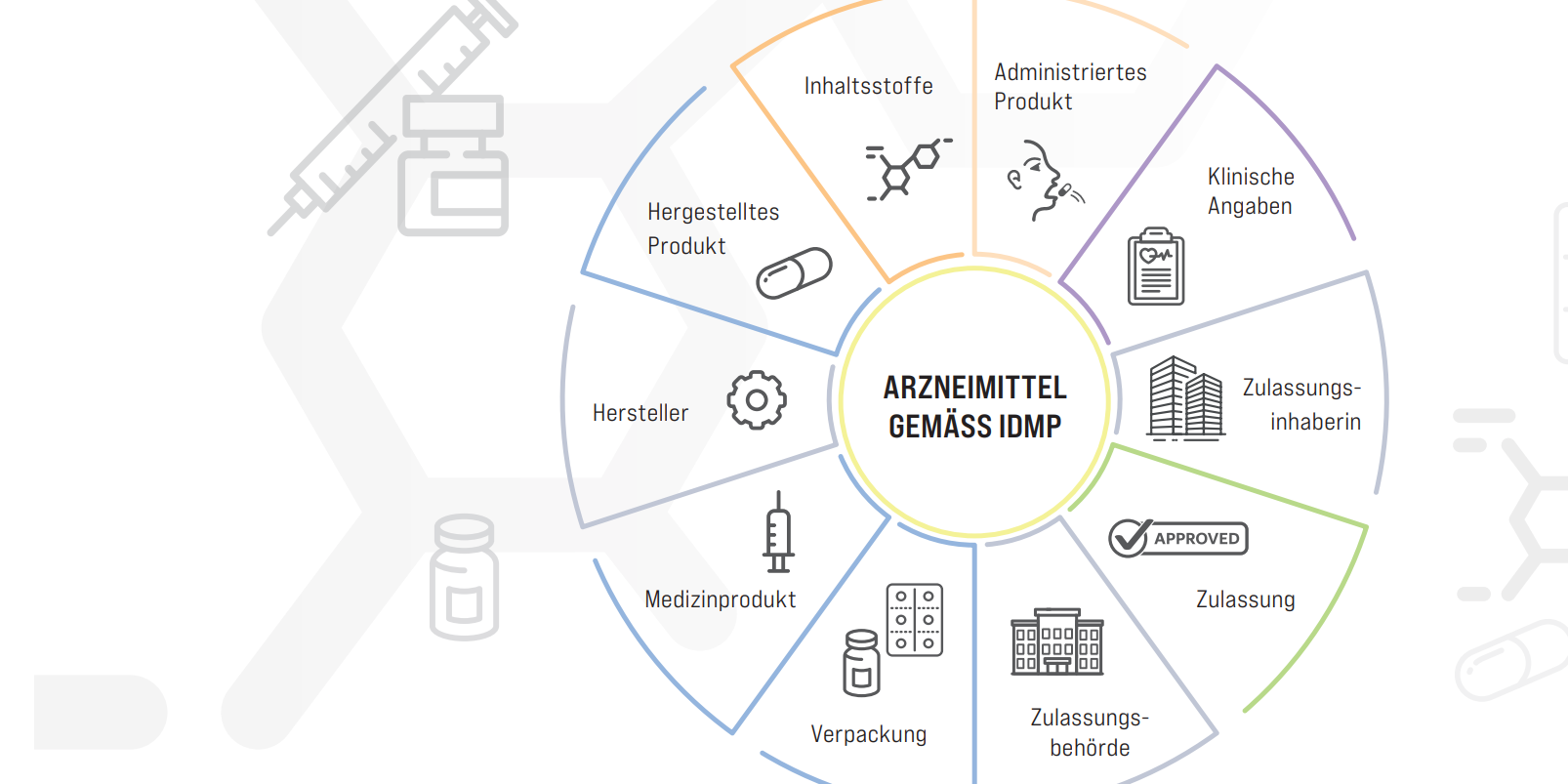
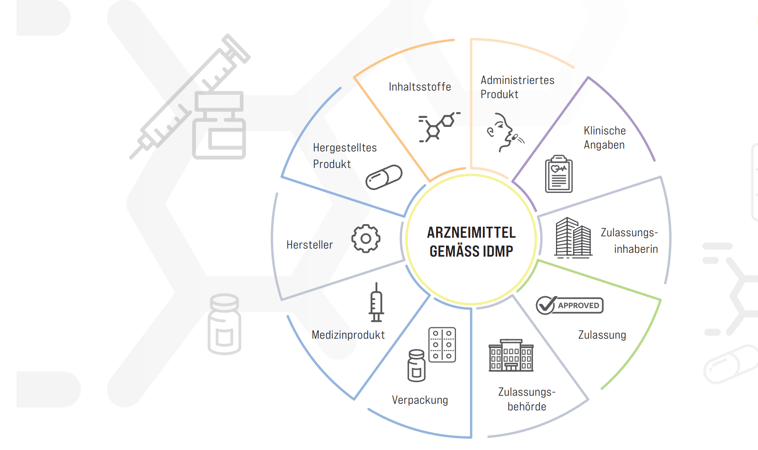
IDMP steht für «Identification of Medicinal Products» und basiert auf fünf dazu geschaffenen ISO-Normen. Sie regeln den Einsatz von strukturierten Daten in der Zulassung, um eine weltweit einheitliche Systematik zur Identifikation und Beschreibung von Arzneimitteln zu ermöglichen. Damit sollen die Patientensicherheit gesteigert und die Effizienz im Datenaustausch zwischen Zulassungsbehörden untereinander und den Firmen verbessert werden. Swissmedic arbeitet seit Jahren in den entsprechenden Arbeitsgruppen mit und beabsichtigt, der EMA mit nur wenigen Jahren Abstand bei der Einführung des neuen Standards zu folgen.
Das Regelwerk legt fest, welche Daten von den Zulassungsinhaberinnen verpflichtend anzuliefern sind: Einheitlich codiert werden z. B. die Inhaltsstoffe, aber auch exakt, wie sich das «Hergestellte Produkt» = Brausetablette vom «Administrierten Produkt» = Trinkflüssigkeit unterscheidet.
Quelle: HealthPoint Magazin 01/22. Die Farben entsprechen dem offiziellen IDMP-Regelwerk.
Das System ist so umfangreich, dass es in mehreren Stufen eingeführt werden soll. In der ersten von vier Einführungsphasen (Iterationen) sollen für die Firmen, bei einer Zulassung durch die Europäische Arzneimittel-Agentur (EMA), 150 Datenelemente verpflichtend werden. Das Architekturdatenmodell von IDMP soll im Endausbau über 500 Datenelemente umfassen und setzt diese in ein sehr komplexes System mit definierten Beziehungen. Zusätzlich zu Angaben, die man erwarten würde, wie Zusammensetzung und Darreichungsform, sollen die Zulassungsinhaberinnen einerseits Indikationen und Nebenwirkungen aus der Rubrik «Klinische Angaben» codieren, andererseits auch präzise Angaben beispielsweise zu den Massen der Verkaufspackung liefern oder in welchen Komponenten das Medikament in einer Packung enthalten ist.
Bringen neue Standards in der Zulassung einen Digitalisierungsschub in die Branche? Ist eventuell nur mit Mehraufwand für Zulassungsinhaberinnen zu rechnen? Die Health Point Redaktion berichtet darüber in der neusten Ausgabe 01/22.



